Tabix
January 13, 2021
If tabix were a character, it would be the swordsman Roronoa Zoro from One Piece, slicing through your genome data to deliver you the result you want.

Tabix is part of Samtools. It indexes a tab-delimted genome
position file (VCF file for example). Once indexed, it can quickly
retrieve data from any part of the file without decompressing it.
Thank you Heng Li 🙏 (tabix author). Here are some things you can
do with tabix to a vcf.gz file.
First, you’ll want to index your VCF like this. This index
acts lke a table of contents, helping tabix jump around the
.gz file quickly.

The index file is called .tbi and will be put
adjacent to your .gz file.

Now you can check for the existence of a variant like so.

Its important to remember, while you’re doing this, that a polymorphism being in the VCF file is not synonymous with being interesting or important or rare. There are lots of harmful / interesting / important polymorphisms in the reference genome. But more on that in another post.
You can look for variants in a region in the same way.
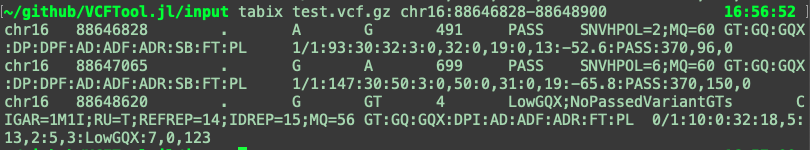
You can also do other useful / cool things with tabix, like return the header of a VCF.
tabix -H vcf.gz
Return chromosome names of a VCF.
tabix -l vcf.gz
You can also return variants found in regions listed in a file. The file can be a bed (.bed, .bed.gz, .bed.bgz) or a TAB-delimited file with CHROM, POS, and, optionally, POS_TO columns.
I like to use BED files. BED files have only 3 required columns:
chromosome, start position, and end position. They have 9 more
optional columns. The only optional column I use is the 4th one,
which is name. I use it because it helps me remember
what that locus is. More on the BED file format
here. Here is a BED file I might use.
chr7 150999022 150999023 rs1799983
chr19 51354483 51354484 rs79338777
chr21 36146407 36146408 rs1056892
NOTE:This bed file is searching for three SNPs.
Notice how the positions are one base apart? The position range in
regions listed in a file need to start one base before where the
expected variant is. That is different from when we search for a
SNP directly like
tabix test.vcf.gz chr7:150999023-150999023 for
example, where the position range is from one position to the same
position. If you were to do this in the BED file and a variant
actually existed at position 150999023 on chromosome 7, it
wouldn’t be returned. I don’t know why this is the
case, but its certainly must-know behavior.
To return variants in regions listed in a file, enter:
tabix -R test.bed vcf.gz
Tabix doesn’t have a stdout option, so you would save the output to a file like this:
tabix -R test.bed vcf.gz > results.tsv
Careful with that .gz file …
gzip and bgzip are not the same thing.
Wait … what is bgzip? Good question.
bgzip is a compression algorithm that creates a
series of gzip compressed blocks which are each 64kb
in size. To go along with all these little blocks is whats called
a “virtual offset”. The vitual offset is actually an
unsigned (meaning nonnegative) 64 bit integer. And this integer
holds information which acts like a map of the
bgzip file. Its because of the “block +
map” structure that bgzip files can be accessed
without unzipping the entire file. And this is why tabix can
access lets say chr1:452923-452923 without unzipping
the whole variants.vcf.gz (a bgzipped file).
But why did I say be careful? Because bgzip files and
gzip files both have the filename extension
.gz. So how do you know if a .gz file is
bgzipped or just gzipped? Because for the reasons explained above,
you cannot access a part of a gzip file without
unzipping it, which is why like tabix requires
bgzip compression. Luckily its easy to tell. Just try
to tabix it (create an index) and if its not bgzipped, tabix will
tell you:

Learn more about bgzip and gzip in
Peter Cock’s detailed post.